УДК 616-056:616.34-008.87
Alterations in Gut Microbiota Composition in Familial
Mediterranean Fever
(Submitted by academician K. G. Karageuzyan 16/IX 2006)
Keywords: familial Mediterranean fever, gut microbiota, phylogenetic analysis Familial Mediterranean fever (FMF; MIM249100) is a
recessively inherited disorder of the inflammatory pathway, manifested by acute
self-limited recurrent episodes of fever and polyserositis [1]. The
Mediterranean fever gene (MEFV), responsible for the disease, has been recently
identified by positional cloning [2, 3]. Pyrin, the protein product of MEFV,
consists of several conserved domains, including the N-terminal pyrin domain (PYD),
which is found in a number of autoinflammatory proteins involved in the
regulation of inflammation and apoptosis [4]. According to recent studies,
autoinflammatory genes, such as MEFV, may represent an exaggerated innate immune
response to various signals in vitro, including microbial products [5]. Indeed,
the CARD15/NOD2 gene product belongs to the same superfamily of proteins [6],
and its mutations have been found to underlie inflammatory bowel diseases (IBD),
such as Crohn's disease, in which an inappropriate immune response to components
of the commensal microbiota exists [7]. In this regard, it has been proposed to
investigate the composition of gut microbiota in FMF to reveal a possible
contribution of commensal bacteria to the onset and maintenance of the disease. Тable 1
As a large majority of bacterial species is effectively
unculturable, it is impossible for detailed examination of gut microorganisms to
be achieved through traditional culture techniques. Molecular-genetic analyses
of bacterial microbiota based on 16S ribosomal ribonucleic acid (rRNA) genes
obviate the need for culture and have been shown to be powerful tools in
determining microbial diversity in complex samples [8].
In
the present study, the fecal bacterial composition has been for the first time
examined in FMF by using microbial community analysis through sequencing of 16S
rDNA libraries.
Fecal samples were collected from
genetically ascertained FMF patients (12 patients in remission, 3 patients in
attack periods) and 7 healthy individuals.
DNA was extracted from fecal samples of FMF patients and healthy
subjects using QIAamp DNA Stool Mini Kit (Qiagen, UK), according to the
manufacturer's instructions. DNA samples were transferred to the Rowett Research
Institute (UK) where 16S rDNA clone libraries were generated, and phylogenetic
analysis was performed. Bacterial 16S rDNA was PCR-amplified with universal
primers covering most intestinal bacterial species (Table 1). The amplicons were
cloned into Escherichia coli chemically competent cells using the pCR-4
TOPO TA Cloning Kit (Invitrogen, UK), according to the manufacturer's
instructions. Recombinant colonies were randomly picked and sequenced on the
automated DNA-sequencer (Beckman, USA) with 926R bacterial primer (Table 1).
Alignment of sequences with reference 16S rDNA gene sequences from healthy gut
microflora was performed using the multiple sequence alignment programme
CLUSTALX v. 1.83 [9]. Phylogenetic analyses were performed using the
neighbor-joining algorithm [10]. Operational taxonomic units (OTUs) were
identified by online Basic Local Alignment Search Tool (BLAST) program at the
NCBI website [11], using search results of at least 99% sequence similarity.
| Application | Primer | Position | Sequences (5'-3') |
| PCR | Fd1 | 8-271 | AGAGTTTGATCMTGGCTCAG |
| PCR | Rp2 | 1492-15101 | ACGGCTACCTTGTTACGACTT |
| Sequencing | 926R | 907-9261 | CCGTCAATTCCTTTGAGTTT |
Using a molecular approach, for the first time, the
composition of fecal microbiota in FMF patients with both inactive and active
stages of the disease, as well as in healthy subjects, was determined. It was
demonstrated that fecal microbiota in FMF differed from that of the healthy
state both in remission and attack periods of the disease.
Three
16S rDNA libraries from fecal samples of FMF remissions, acute FMF and healthy
controls were generated. A total of 1328 clones (572 for healthy controls, 629
for FMF remission and 127 for FMF attack) were analyzed, and phylogenetic
relationships of main bacterial phyla in each studied group were established
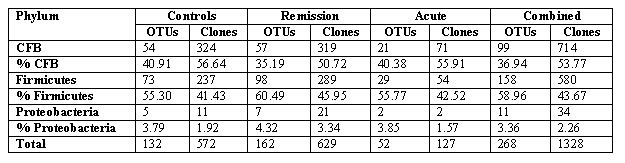
(Fig. 1 A, B, C). Among the 1328 clones analyzed, there were 268 distinct OTUs,
which fell into three major phyla: Cytophaga-flavobacter-bacteroides (CFB)
group, Firmicutes, and Proteobacteria. The overall distribution of
the three dominant bacterial phyla among the three subsets of subjects is shown
in Table 2.
|
|
Тable 2
|
|
As shown in Table 2, Bacteroides was the most
abundant group in all three cohorts, followed by the Firmicutes. The
relative proportions of CFB and Firmicutes were not markedly different
among the three groups; however, significant differences were detected in
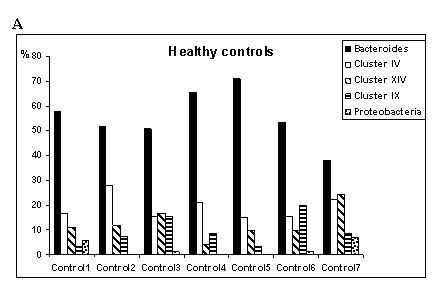
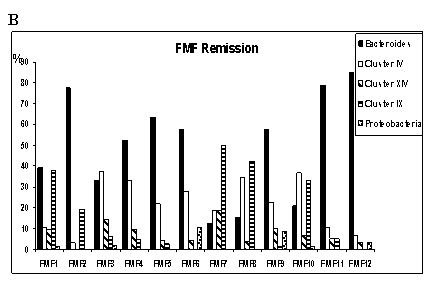
bacterial subgroups within these main phyla (Fig. 1A, B, C). In Fig. 1B groups
of FMF patients in remission were determined according to the biodiversity in
the main phylogenetic groups, demonstrating high variability, in contrast to
stable composition of gut bacteria in healthy state (Fig. 1A). Particularly,
there is a group (FMF2, FMF4, FMF5, FMF6, FMF9, FMF11, and FMF12)
overrepresented by OTUs belonging to the CFB phylum, which amounted up to 50-55%
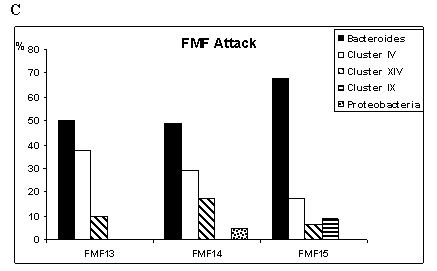
of gut bacteria in healthy subjects (Fig. 1A) and acute FMF (Fig. 1C). In the
second group of FMF remissions (FMF1, FMF7, FMF8, and FMF10) there is a
substantially higher proportion of cluster IX of Propionate-producing bacteria,
as compared to healthy controls. Interestingly, these bacteria tended to
disappear during the attack period (Fig. 1C). The pairwise comparisons of each
16S rDNA library to every other library also revealed significant alterations in
gut microbiota composition in FMF compared to the norm (Table 3). In particular,
the Prevotellaceae subgroup (within CFB) was significantly low in active
stage of FMF as compared to FMF remission and healthy state (16.5%, 22% and
27.6%, respectively), in contrast to Bacteroidaceae (within CFB) subgroup
(30.7%, 17.8% and 21.7%, acute, remission and healthy, respectively). The
Butyrate-producing Faecalibacterium group was higher in active FMF
compared to both FMF remissions and controls (14.2% in attack vs. 6.5%).
Gamma-proteobacteria were 0.2% and 2.1%, in healthy controls and FMF
remission, respectively, and there was a complete loss of these bacteria in the
acute phase. The most striking difference was observed in the
Propionate-producing Acidaminococcaceae subgroup (Clostridial cluster IX
within Firmicutes). These bacteria were overrepresented in remission
period compared to controls (16% vs. 10%), and tended to disappear in attack
(3%), found only in FMF15 (Fig. 1C). Although in the latter group the bacterial
sequences were the least diverse, which might be the consequence of a general
inflammatory process, however representatives of the Butyrate-producing
Faecalibacterium group in attack were significantly high compared to both
FMF remission and healthy state (Table 3). Butyrate, which is produced by
bacterial fermentation, has been shown to reduce inflammation in experimental
colitis in animal models. It reduces inflammation through an inhibitory effect
on proinflammatory cytokine expression, thus demonstrating anti-inflammatory
properties [12]. Such increase of butyrate producers among acute patients
implies that it could correspond to a compensative response.
Тable 3
| Bacterial subgroups | Healthy | Remission | Attack |
| Prevotellaceae (CFB)* | 27.6% | 22% | 16.5% |
| Bacteroidaceae (CFB)** | 21.7% | 17.8% | 30.7% |
| Faecalibacterium (Cluster IV)*,**,*** | 6.5% | 6.5% | 14.2% |
| Acidaminococcaceae (Cluster IX)*,**,*** | 10% | 16% | 3% |
| Gamma-proteobacteria*** | 0.2% | 2.1% | 0 |
We observed no specific microbial group pointing to the presence of bacteria, which could be specifically involved in disease activity. The 16S rDNA profile of the fecal microbiota was very stable under healthy conditions but unstable in FMF patients. It seems the alterations in gut microflora composition reflect a metabolic imbalance of the complex microbial ecosystem with severe consequences for the host immune system. How some bacteria may exert an inflammatory effect and others a protective role in FMF is yet uncertain. Is a breakdown in the balance between putative "protective" and "harmful" intestinal bacteria simply a secondary phenomenon in FMF, or is altered composition a primary modification, that is to say genetically determined, leading to an inflammatory process? Further studies may help to explain the complex relationships among bacteria, inflammation and genetics, which could provide new insights into the pathogenesis and treatment of FMF.
Institute of Molecular Biology NAS RA
1. Sohar E., Gafni J., Pras M., Heller H. -
Am. J. Med. 1967. V. 43. P. 227-253.
2. The International FMF Consortium. - Cell. 1997. V. 90.
P. 797-807.
3. The French FMF Consortium. -
Nat. Genet. 1997. V. 17. P. 25-31.
4. Dowds T. A., Masumoto J., Chen F. F., Ogura Y., Inohara N., Nunez G.
- Biochem. Biophys. Res. Commun. 2003. V. 302. P. 575-580.
5. Shoham N. G., Centola M., Mansfield E.,
Hull K. M., Wood G., Wise C. A., Kastner D. L. - PNAS. 2003. V.
100. P. 13501-13506.
6. Ogura Y., Inohara
N., Benito A., Chen F. F., Yamaoka S., Nunez G. - J. Biol. Chem.
2001. V. 276. P. 4812-4818.
7. Ogura Y., Bonen D. K., Inohara N., Nicolae D. L., Chen F. F., Ramos R., Britton H., Moran
T., Duerr R. H., Achkar J. P., Brant S. R., Bayless T. M., Kirschner B. S.,
Hanauer S. B., Nunez G., Cho J. H. - Nature. 2001. V. 411. P.
603-606.
8. Eckburg P. B., Bik E. M.,
Bernstein C. N., Purdom E., Dethlefsen L., Sargent M., Gill S. R., Nelson K. E.,
Relman D. A. - Science. 2005. V. 308. P. 1635-1638.
9. Thompson J. D., Gibson T. J., Plewniak
F., Jeanmougin F., Higgins D. G. - Nucleic Acids Research. 1997.
V. 24. P. 4876-4882.
10. Saitou N., Nei M.
- Mol. Biol. Evol. 1987. V. 4. P. 406-425.
11. http://www.ncbi.nlm.nih.gov/BLAST/
12.
Segain J. P., Raingeard
Bletiere D., Bourreille A., Leray V., Gervois N., Rosales C., Ferrier L., Bonnet
C., Blottiere H. M., Galmiche J. P. - Gut. 2000. V. 47. P.
397-403.