УДК 577.155.3, 577.112.4
Инактивация аргиназы гидроксиламином
(Представлено академиком М.А. Давтяном 10/VI 2004)
Аргиназа (К 3.5.3.1.) катализирует
расщепление аргинина на мочевину и орнитин. Кроме участия в орнитиновом цикле,
аргиназа, как известно, играет важную роль и во многих других процессах,
происходящих в различных тканях живых организмов [1,2]. В последние годы интерес
к аргиназе был стимулирован демонстрацией ее участия в метаболизме окиси азота
[3,4].
|
|
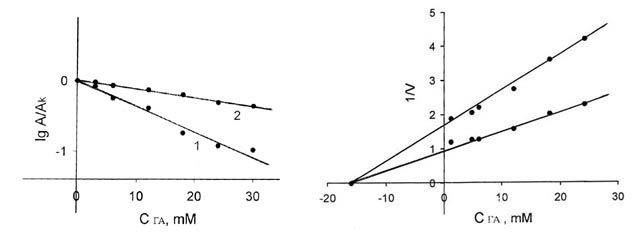
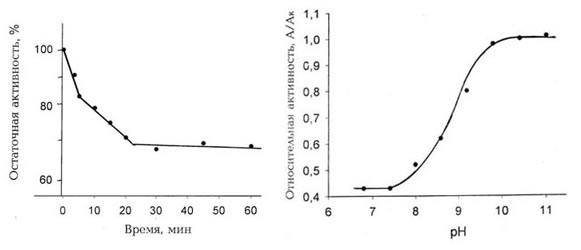
Инактивация аргиназы при взаимодействии с ГА
происходит в первые 20 мин, и дальнейшая инкубация почти не влияет на активность
фермента. На рис. 1,А показана кинетика инактивации аргиназы в присутствии
10 mM ГА при рН 7.6. Как видно из рисунка, инактивация фермента представляет
собой двустадийный процесс, каждая из стадий которого протекает по реакции
псевдопервого порядка. Это означает, что в ходе реакции с ГА два типа
чувствительных к реагенту функциональных групп фермента модифицируются с
различными скоростями. Взаимодействующие с реагентом карбоксильные или
карбонильные группы, находящиеся в активированном состоянии, возможно,
расположены вблизи от активного центра аргиназы или входят в его состав.
Активация карбоксильных групп происходит, по-видимому, за счет взаимодействия с
другими функциональными группами белковой молекулы, находящимися в
непосредственной близости [14].
|
|
Для проверки влияния модификации ГА на
конформационное состояние молекулы аргиназы были измерены спектры ФЛ растворов
фермента до и после воздействия реагента. Инактивацию аргиназы проводили при рН
6 (0.05 M фосфатный буфер) и концентрации ГА 48 mM в растворе в течение 60 мин
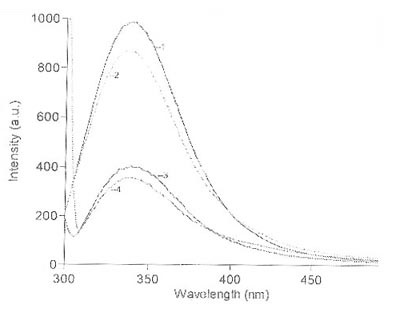
при комнатной температуре. Максимум спектра ФЛ исследуемого препарата аргиназы
при длине волны возбуждения (lвозб) 297 нм находится при 338 нм, что было показано ранее
[18,19]. При длине волны возбуждения 280 нм интенсивность ФЛ значительно
возрастает, а максимум ФЛ смещается в коротковолновую область до 336-337 нм.
Небольшое смещение положения максимума ФЛ в коротковолновую область указывает на
то, что флуоресценция остатков тирозина, очевидно, вносит определенный вклад в
процессы ФЛ аргиназы, однако спектр ФЛ при lвозб 280 нм, в основном, определяется триптофановой
компонентой. Согласно литературным данным [20], число остатков тирозина на
молекулу аргиназы печени быка в 3.5 раза больше, чем остатков триптофана.
Возможно, в данных условиях в молекуле аргиназы происходит миграция энергии с
остатков тирозина на триптофан. Необходимо также учитывать, что в белках имеет
место тушение флуоресценции находящимися в непосредственной близости от
хромофоров группами, обладающими тушащими свойствами, среди которых аминогруппы,
карбоксильные группы, имидазольная группа гистидина и т.п. [21]. Отношение
интенсивностей ФЛ в максимуме при длинах волн возбуждения 280 и 297 нм
(![]() ) составляет 2.48.
Отношение относительных квантовых выходов ФЛ аргиназы
(q280/q297) при указанных длинах волн возбуждения равно
2.3.
) составляет 2.48.
Отношение относительных квантовых выходов ФЛ аргиназы
(q280/q297) при указанных длинах волн возбуждения равно
2.3.
|
|
В результате взаимодействия с ГА
интенсивность ФЛ растворов аргиназы для двух длин волн возбуждения снижается
одинаково - на 11.5%, однако положение максимумов и отношение максимальных
интенсивностей ФЛ не меняется,
Ереванский государственный университет
Литература 1. Reddi S.R.R., Campbell J. W.
- Biochem. J. 1969. V.115. N 3. P.
305-314.
![]() = 2.47 (рис. 4).
Несколько уменьшается лишь q280 / q297 = 2.2. Таким образом,
параметры ФЛ растворов аргиназы существенно не меняются, т. е. изменений
микроокружения остатков триптофана и тирозина аргиназы, согласно полученным
данным, в этих условиях не происходит. Снижение интенсивности ФЛ аргиназы в
результате взаимодействия с ГА можно объяснить увеличением числа тушащих групп
на поверхности молекулы белка. При модификации гидроксиламином, возможно, имеют
место некоторые изменения конформации в области активного центра аргиназы,
однако они не затрагивают ту часть молекулы белка, в которой расположены остатки
тирозина и триптофана. Полученные данные еще раз подтверждают наличие в составе
этого фермента устойчивых к различным воздействиям гидрофобных участков, в
которых аминокислотные остатки связаны друг с другом прочными связями,
поддерживающими вторичную и третичную структуру, что обеспечивает высокую
стабильность молекулы аргиназы в организме в различных экстремальных условиях.
= 2.47 (рис. 4).
Несколько уменьшается лишь q280 / q297 = 2.2. Таким образом,
параметры ФЛ растворов аргиназы существенно не меняются, т. е. изменений
микроокружения остатков триптофана и тирозина аргиназы, согласно полученным
данным, в этих условиях не происходит. Снижение интенсивности ФЛ аргиназы в
результате взаимодействия с ГА можно объяснить увеличением числа тушащих групп
на поверхности молекулы белка. При модификации гидроксиламином, возможно, имеют
место некоторые изменения конформации в области активного центра аргиназы,
однако они не затрагивают ту часть молекулы белка, в которой расположены остатки
тирозина и триптофана. Полученные данные еще раз подтверждают наличие в составе
этого фермента устойчивых к различным воздействиям гидрофобных участков, в
которых аминокислотные остатки связаны друг с другом прочными связями,
поддерживающими вторичную и третичную структуру, что обеспечивает высокую
стабильность молекулы аргиназы в организме в различных экстремальных условиях.
2. Schrell A.,
Alt-Moerbe J., Lanz T., Schroeder J. - Eur. J. Biochem.
1989. V. 184. P. 635-641.
3. Daghigh F., Fukuto J. M., Ash D.E. - B.B.R.C. 1994.
V. 202. N 1. P. 174-180.
4. Chang Ch-I., Liao J. C., Kuo L. - Amer. J. Physiol.
1998. V. 274. N 1. Pt 2. P. 342-348.
5. Perozich J., Hempel J., Morris S.M. Jr. -
B.B.A. 1998. V. 1382. N 1. P. 23-37.
6. Muszynska G., Severina L.O., Lobireva L.W. -
Acta biochim. Polon. 1972. V.19. N 2. P.
109-116.
7. Carvajal N., Uribe
E., Salas M. - Biochem. Arch. 1966. V. 12. N 1.
P. 19-26.
8. Давтян М. А.,
Геворкян М. Л. - Ученые записки ЕГУ. 1997. N 1. С.
40-47.
9. Ber E., Muszynska G.
- Acta biochim. Polon. 1979. V. 26. N 1-2.
P. 103-114.
10. Daghigh F.,
Cavalli R.C., Soprano D.R., Ash D.E. - Arch. Biochem.
Biophys. 1996. V. 327. N 1. P. 107-112.
11. Kanyo Z.F. Scolnick L.R., Ash D.E., Christianson D.W. - Nature. 1996. V. 383. N 6600. P.
554-557.
12. Геворкян М. Л.,
Давтян М. А. - Ученые записки ЕГУ. 2000. N 2.
С. 79-82.
13. Гончар Н. А.,
Мардашев С. Р. - Биохимия. 1970. Т. 35. N 2.
С. 224-228.
14. Аваева С.
М., Воробьева Н. Н., Мельник М. С., Назарова Т. И. - Биоорг.
химия. 1979. Т. 5. N 10. С. 1570-1578.
15. Balandin T., Fernandez V. M., Aparicio P.J. -
Plant Physiol. 1986. V. 82. N 1.
65-70.
16. Jencks W.P.
- J. Am. Chem. Soc. 1958. V. 80. N 17. P.
4585-4588.
17. Hosoyama Y.
- Eur. J. Biochem. 1972. V. 27. N 1. P.
1675-1677.
18. Геворкян М. Л.,
Давтян М. А. - Ученые записки ЕГУ. 2001. N 1.
С. 100-105.
19. Burstein E.
R., Vedenkina N.S., Ivkova M. N. - Photochem. Photobiol.
1973. V. 18. P. 263-279.
20. Harrel D., Sokolovski M. - Eur. J. Biochem. 1972. V.
25. N 1. P. 102-108.
21. Бурштейн Э.А. Молекулярная биология. 1983. Т. 17. N
3. С. 455-465.